Connor Scott
Scientist (Alkermes) / Honorary Research Fellow (Oxford)
Alkermes
Nuffield Department of Clinical Neurosciences
Professional Profile
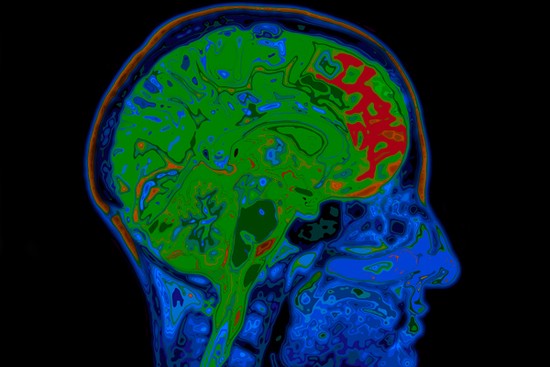
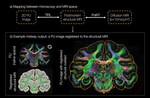
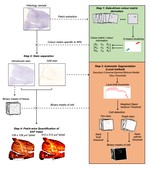
Connor Scott is an experienced neuroscientist with over 12 years of expertise in ex vivo research, specialising in the analysis of human and rodent tissue, histology, molecular biology, and mass spectrometry.
Throughout his career, Connor has been dedicated to advancing our understanding of the molecular mechanisms underlying neurodegenerative and psychiatric disorders. His work spans cutting-edge research initiatives and innovative drug discovery programs aimed at uncovering novel therapeutic solutions.
Connor is a recognised subject matter expert in:
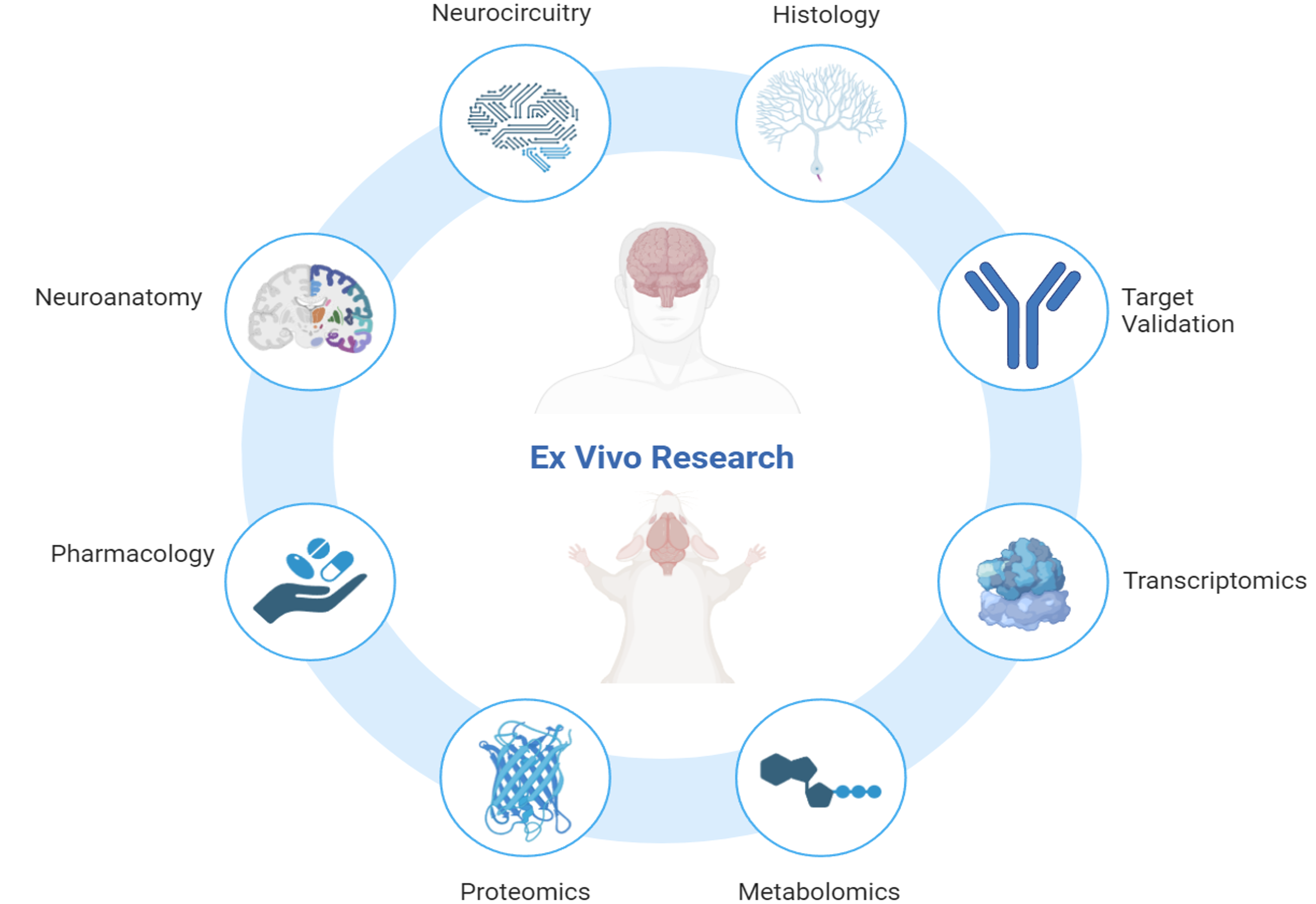
Connor’s research encompasses some of the most challenging and impactful areas of neuroscience, including neurodegenerative conditions such as Alzheimer’s disease, frontotemporal dementia, and amyotrophic lateral sclerosis (ALS). He also brings significant expertise to psychiatric disorders like schizophrenia and complex diseases with neurological components such as narcolepsy and glioblastoma multiforme.
With a passion for advancing neuroscience and a track record of impactful contributions, Connor continues to drive innovation at the intersection of research and drug discovery.
Scroll down to see a overview of his professional contributions or click on the headings above to see a detailed overview.
Interests
- Drug Discovery / Drug Development
- Cell Biology / Molecular Biology
- Histology / Neuropathology
- Neurodegeneration / Psychiatry
- Proteomics / Transciptomics
Education
D.Phil/PhD in Clinical Neurosciences, 2021
University of Oxford
BSc (Hons) in Biomedical Sciences, 2012
University of Greenwich