Quantitative patterns of motor cortex proteinopathy across ALS genotypes
Abstract
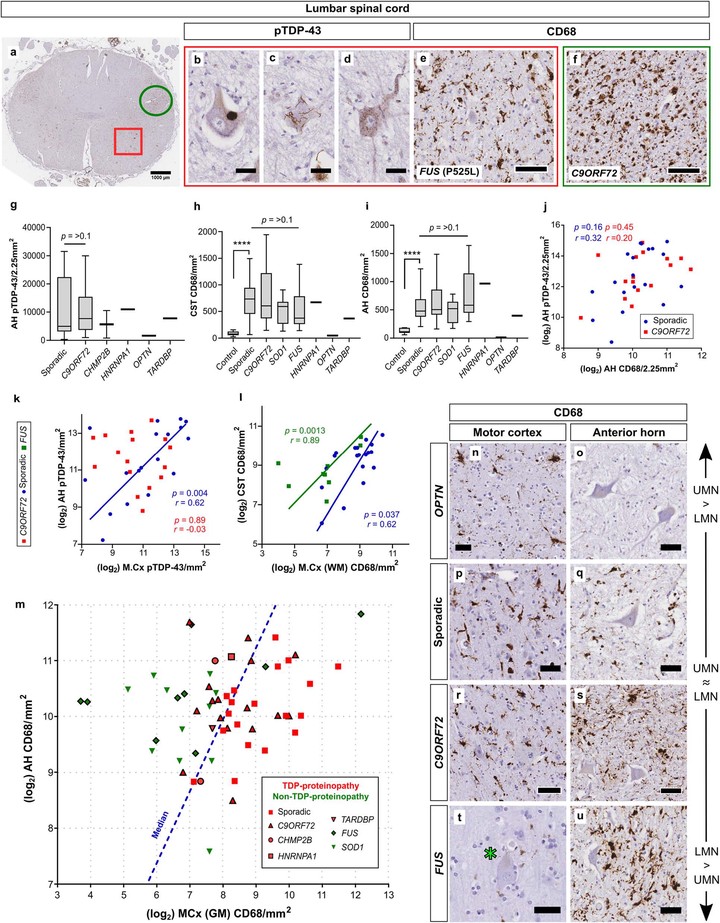
Degeneration of the primary motor cortex is a defining feature of amyotrophic lateral sclerosis (ALS), which is associated with the accumulation of microscopic protein aggregates in neurons and glia. However, little is known about the quantitative burden and pattern of motor cortex proteinopathies across ALS genotypes. We combined quantitative digital image analysis with multi-level generalized linear modelling in an independent cohort of 82 ALS cases to explore the relationship between genotype, total proteinopathy load and cellular vulnerability to aggregate formation.
Type
Publication
Acta Neuropathologica Communications